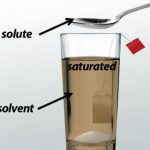
Question 1 What is a saturated solution? Explain with example? Question 2 What is the effect of temperature on saturated solution? Question 3 Define the term solubility. Give example? Also Read NCERT Solutions for Chapter 5 Separation of Substances When we dissolve a substance in water then a solution is formed. A given quantity of water can dissolve only a certain … [Read more...] about Saturated Solution
saturated solution
Concentration
Question 1 What is meant by the concentration of a solution? Question 2 How much water should be added to 15g of salt to obtain 15 per cent salt solution? Question 3 Calculate the concentration of a solution which contains 2.5g of salt dissolved in 50g of water? Question 4 What does a 15% alcohol solution means? Question 5 If 25mL of acetone is present in 150ml of … [Read more...] about Concentration